Ziren Research aims to develop new, effective cancer therapeutics.
Past and present cancer therapeutics:
- Fail to treat cancers diagnosed at late stages, due to failure in stopping cancer recurrence in the original or distance site.
- Aim to kill cancer cells by chemo-, radiation- and/or targeted-therapies to block growth-promoting signaling pathways, etc. This doesn’t work.
- Aim to starve cancer by anti-angiogenesis therapy – this has also failed.
Why can’t cancer treatment succeed by simply target angiogenesis? Cancer cells can adopt a vascular state to form vasculogenic mimicry.
Why can’t cancer treatment succeed by simply targeting angiogenesis? Cancer cells survive at higher rates than normal cells in poor conditions like environments low in oxygen and nutrient supply or high in acidity. Such conditions, in fact, trigger their transition to a vascular state, allowing blood vessels to form within tumor.
Why can’t cancer treatment succeed by blocking one or two growth-promoting signaling pathways? Cancer quickly develops drug-resistance by altering growth-promoting mechanisms.
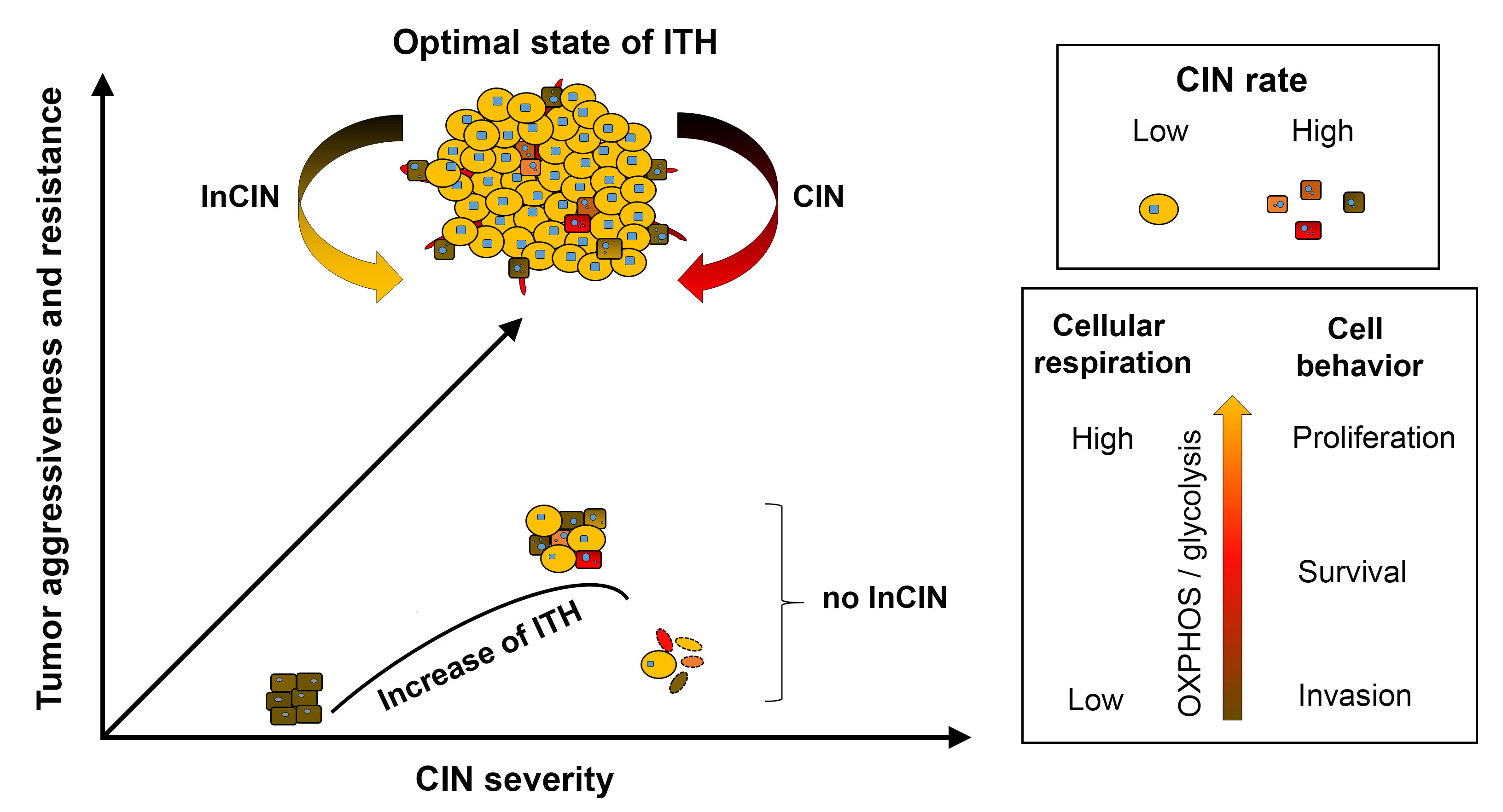
Why has targeting cancer stem cells failed? Cancer cells, including cancer stem cells, are changeable due to their unstable genome, namely chromosomal instability (CIN). Cancer is formed by populations differentially responsible for the intra-tumoral heterogeneity (ITH), including those functional responsible for the “growing” and “going” features.
Recruiting the body’s natural defense to fight cancer with immunotherapy has shown promise. However, limitations arise due to major similarities between normal cells and cancer cells, especially cancer stem cells, with little differences of their surface antigens.
Cancer’s Evolutionary Tactics
- Control of chromosome instability rate and modulating intra-tumoral heterogeneity
- Cancer-specific metabolism and energetics
Along the long path of cancer treatment, our failure in curing cancer necessitates rethinking our strategy for future cancer therapeutics. The tactics utilized in cancer evolution would persist and manifest in the recurrence of cancer after standard therapy, including surgical removal of tumor, which remains the most effective treatment for most patients with solid cancer.
Instead of relying on current treatment models such as surgical removal, we can look to using cancer’s own evolutionary mechanisms against itself. Exploiting cancer’s tactics could turn cancer from a devastating death sentence to a manageable chronic disease.
|
|
The cancer evolutionary tactic of controlling and optimizing the state of ITH. In the late steps of cancer evolution, neo-transformed cells become different functional components of a tumor, which defines intra-tumoral heterogeneity (ITH). The common metabolic feature of different tumor subpopulations is using glycolysis on top of oxidative phosphorylation (OXPHOS). The common genomic feature of tumor cells isaneuploidy. Tumor ecology streamlines tumor subpopulations to the essential ones, leading to optimal state of ITH. This leads to aggressive tumor behavior and resistance to adverse microenvironmental factors. CIN speeds up cancer evolution by increasing ITH, while inhibition of CIN (InCIN) maintains the optimal state of ITH. This figure is shown in Cancers 2020, 12(6), 1649; https://doi.org/10.3390/cancers12061649 |
Weaponizing human EGF-containing fibulin-like extracellular protein 1 (EFEMP1) for 21st century cancer therapeutics, a published study by Zhou et al. in Oncoscience 2016 May 23; 3 (7-8): 208-219
In cancer, proteins or microRNAs with dual roles can differentially modulate cancer cells different in karyotype and metabolism to orchestrate and balance a tumor’s opposing and balanced “grow” and “go” behaviors in response to changes in local microenvironments, such as deprivation of nutrients and oxygen. Fibulin-3 (official name EFEMP1) is an extracellular protein with a dual role in cancer. Hypermethylation of EFEMP1 promoter and suppression of EFEMP1 expression have been reported in majority types of cancer with local aggressive growth, such as breast, colon, lung, liver, nasopharynx, and prostate [1-7], while upregulated expression of EFEMP1 was also reported in cervical carcinoma with lymph node metastasis [8]. The anti-tumor effects of EFEMP1 are most likely accounted for by its anti-angiogenic effect [9], its anti-EGFR/AKT-mediated growth signaling effect [5, 10-12], and its anti-IGF1R [13] and/or anti-TGF-beta [14] mediated epithelial-to-mesenchymal transition effects, as well as its function in normal tissue to inhibit the expression and activities of matrix metalloproteinase [15]. EFEMP1’s oncogenic effect has been related to its pro-invasive function in activation of NOTCH [16] and/or NK-kappa B signaling and MMP2 [17]. EFEMP1 could be one of the proteins employed by cancer in its initiation and progression via control of two fundamental pillars of cancer biology, namely chromosome instability (CIN) and intra-tumoral heterogeneity (ITH), by its function to inhibit CIN [18]. A study examining EFEMP1 in tumor mass-forming cells (TMC, the “grower”) and stem-like tumor-initiating cells (STIC, the “goer”) within a GBM cell line U251 and its syngeneic neural sphere culture line U251-NS demonstrated EFEMP1’s context-dependent dual role [19]. This appears to be a tactic of cancer evolution, with the downregulation of its expression in the “grower” population where EFEMP1 inhibits the cell growth by attenuation of EGFR/AKT signaling, and the upregulation of its expression in the “goer” population where EFEMP1 promotes cell invasion by enhancing NOTCH/AKT signaling. Consistently, in gliomas where EFEMP1 expression – and thus the anti-EGFR effect of EFEMP1 – was suppressed, heightened levels of EGFR expression were associated with unfavorable patient outcomes in prognostic models [12]. Hence, simple restoration or elimination of EFEMP1 as a treatment of GBM will fail.
The strategy of dissection and characterization of functional domains and residues of EFEMP1 for its dual function in GBM led to the discovery a human fibulin-3 variant (hF3v), expressed by the EFEMP1 variant construct E5 [20]. Both lentiviral-mediated expression of hF3v and direct administration of ZR30, a protein-form of hF3v made by an in vitro system, showed the anti-EGFR function to be stronger than EFEMP1. They also demonstrated tumor suppression functions EFEMP1 doesn’t have, including lowering the cancer cells’ invasiveness via decreasing the level of NOTCH/AKT signaling and extracellular MMP2 activation, as shown in multiple cell lines of GBM (including two short-term cultured primary cultures), with an anti-MMP2 function also shown in cell lines of prostate and cervix [20, 21]. The study of ZR30 in GBM primary cultures and in vivo models for tumor vascularization and angiogenesis demonstrated the anti-tumor vascularization function of hF3v, via blocking upregulation of transcription factors involved in transdifferentiate tumor cells into vascular state and suppression of angiogenesis [22]. Therapeutic efficacy of the protein-form of hF3v was shown by one dose of the intra-tumoral injection ZR30, which significantly improved survival of nude mice baring an intracranial (i.c.) xenograft of human GBM [20]. That study also showed a 3 and 6-week tumor suppression effect (or in vivo lifespan) of ZR30 in two GBM xenograft models with high and low tumorigenicity, respectively. The coupling of a viral form of hF3v, such as recombinant adeno-associate (AAV) that could transduce both tumor (dividing) and normal (non-dividing) cells around the tumor, with the protein-form of hF3v, delivered into the tumor site after surgical removal of tumor, would establish an immediate and long-last tumor suppressing microenvironment via the extracellular functions of hF3v in order to inhibit formation of recurrent tumor by infiltrated tumor cells. Inhibiting tumor recurrence will significantly help cancer patients. This is the type of novel cancer therapeutic that will work towards making cancer a chronic disease.
REFERENCES
1. Yue, W., et al., Frequent inactivation of RAMP2, EFEMP1 and Dutt1 in lung cancer by promoter hypermethylation. Clin Cancer Res, 2007. 13(15 Pt 1): p. 4336-44.
2. Nomoto, S., et al., Epidermal growth factor-containing fibulin-like extracellular matrix protein 1, EFEMP1, a novel tumor-suppressor gene detected in hepatocellular carcinoma using double combination array analysis. Ann Surg Oncol, 2010. 17(3): p. 923-32.
3. Sadr-Nabavi, A., et al., Decreased expression of angiogenesis antagonist EFEMP1 in sporadic breast cancer is caused by aberrant promoter methylation and points to an impact of EFEMP1 as molecular biomarker. Int J Cancer, 2009. 124(7): p. 1727-35.
4. Kim, Y.J., et al., EFEMP1 as a Novel DNA Methylation Marker for Prostate Cancer: Array-Based DNA Methylation and Expression Profiling. Clin Cancer Res, 2011. 17(13): p. 4523-30.
5. Hwang, C.F., et al., Fibulin-3 is associated with tumour progression and a poor prognosis in nasopharyngeal carcinomas and inhibits cell migration and invasion via suppressed AKT activity. J Pathol, 2010. 222(4): p. 367-79.
6. Tong, J.D., et al., Downregulation of fibulin-3 gene by promoter methylation in colorectal cancer predicts adverse prognosis. Neoplasma, 2011. 58(5): p. 441-8.
7. Wang, R., Y.W. Zhang, and L.B. Chen, Aberrant promoter methylation of FBLN-3 gene and clinicopathological significance in non-small cell lung carcinoma. Lung Cancer, 2010. 69(2): p. 239-44.
8. En-lin, S., C. Sheng-guo, and W. Hua-qiao, The expression of EFEMP1 in cervical carcinoma and its relationship with prognosis. Gynecol Oncol, 2010. 117(3): p. 417-22.
9. Albig, A.R., J.R. Neil, and W.P. Schiemann, Fibulins 3 and 5 antagonize tumor angiogenesis in vivo. Cancer Res, 2006. 66(5): p. 2621-9.
10. Hu, Y., et al., EFEMP1 suppresses malignant glioma growth and exerts its action within the tumor extracellular compartment. Mol Cancer, 2011. 10: p. 123.
11. Kim, E.J., et al., Fibulin-3 promoter methylation alters the invasive behavior of non-small cell lung cancer cell lines via MMP-7 and MMP-2 regulation. Int J Oncol, 2012. 40(2): p. 402-8.
12. Hu, Y., et al., Anti-EGFR function of EFEMP1 in glioma cells and patient prognosis. Oncoscience, 2014. 1(3): p. 205-15.
13. Kim, I.G., et al., Fibulin-3-mediated inhibition of epithelial-to-mesenchymal transition and self-renewal of ALDH+ lung cancer stem cells through IGF1R signaling. Oncogene, 2014. 33(30): p. 3908-17.
14. Tian, H., et al., Fibulin-3 is a novel TGF-beta pathway inhibitor in the breast cancer microenvironment. Oncogene, 2015. 34(45): p. 5635-47.
15. Rahn, D.D., et al., Failure of pelvic organ support in mice deficient in fibulin-3. Am J Pathol, 2009. 174(1): p. 206-15.
16. Hu, B., et al., Fibulin-3 promotes glioma growth and resistance through a novel paracrine regulation of Notch signaling. Cancer Res, 2012.
17. Wang, Z., et al., EFEMP1 promotes the migration and invasion of osteosarcoma via MMP-2 with induction by AEG-1 via NF-kappaB signaling pathway. Oncotarget, 2015. 6(16): p. 14191-208.
18. Zhou, Y.H., K. Afrasiabi, and M.E. Linskey, Extracellular control of chromosomal instability and maintenance of intra-tumoral heterogeneity. Journal of Cancer Metastasis and Treatment, 2018. 4(41): p. 15.
19. Hu, Y., et al., Cell context-dependent dual effects of EFEMP1 stabilizes subpopulation equilibrium in responding to changes of in vivo growth environment. Oncotarget, 2015. 6(31): p. 30762-72.
20. Zhou, Y.H., et al., Weaponizing human EGF-containing fibulin-like extracellular matrix protein 1 (EFEMP1) for 21st century cancer therapeutics. Oncoscience, 2016. 3(7-8): p. 208-219.
21. Li, Y., et al., Human fibulin-3 protein variant expresses anti-cancer effects in the malignant glioma extracellular compartment in intracranial xenograft models. Oncotarget, 2017. 8(63): p. 106311-106323.
22. Ke, C., et al., Dual antivascular function of human fibulin-3 variant, a potential new drug discovery strategy for glioblastoma. Cancer Sci, 2020. 111(3): p. 940-950.